



Motoneuron diseases
Our team focuses on the study and treatment of diseases affecting the communication between motor neurons and their muscle target. Using human pluripotent stem cells carrying causative mutations, we are developing new models for various neuromuscular diseases, such as Myotonia Dystrophy Type 1 or Infantile Spinal Muscular Atrophy.
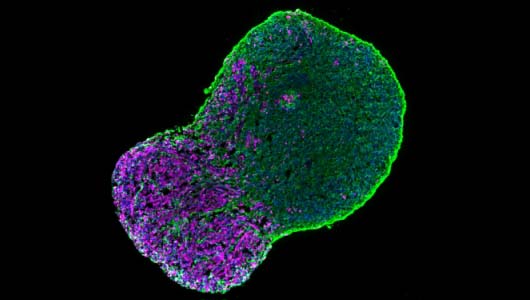
Our team aims to better understand the molecular and cellular mechanisms involved in the development of diseases affecting the motor neuron and their communication with the muscle target and consequently identify new therapeutic avenues. Our research axes are declined around :
1. The generation of new models from human pluripotent stem cells to better understand the communication between motor neurons and their muscle targets. In recent years our group has developed several protocols to efficiently convert human pluripotent stem cells into different types of motor neurons. We have also established several protocols for co-culturing with muscle cells to functionally measure the communication between these two cell types.
We are now working on the establishment of 3D organoids to generate more complex models of the neuromuscular junction.
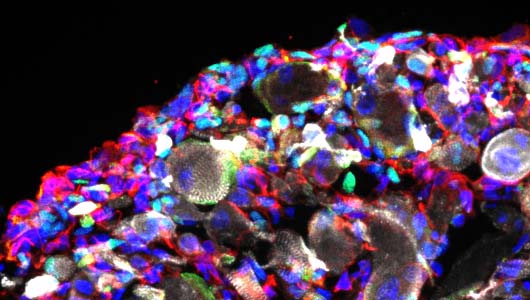
2. Deepening our knowledge of the physiopathological mechanisms involved in certain neuromuscular diseases of genetic origin. Our group is mainly interested in Dystrophic Myotonia Type 1, infantile spinal muscular atrophy and amyotrophic lateral sclerosis.
For this mechanistic research, we are interested in identifying mechanisms that may explain the degeneration or malfunction of these intercellular structures. We are also interested in understanding why certain groups of motor neurons appear to be more susceptible to certain pathological processes. Our final goal is to identify cellular abnormalities that will allow us to identify new therapeutic leads.
3. Our third research axis aims at identifying new therapeutic leads, mainly through pharmacological approaches. Our approaches are essentially based on High Content Screening techniques associated with Machine Learning.
Wolfram’s syndrome
Team members
Cécile Martinat
Team leader and Director of the INSERM unit (U861)
Sandrine Baghdoyan
Research Engineer (Inserm)
Member of the Motoneurone team since its creation, Sandrine studies the in vitro therapeutic potential of repositioned pharmacological compounds as candidates for DM1.
Azania Abatan
Doctoral student
Integrated in the Motoneurone team as part of her M2 internship in February 2021, then as a study engineer in October 2021, Azania started her PhD in January 2023. She is working on the development and characterization of DM1 cortical organoids.
Morgan Gazzola
Post Doctorate (Inserm)
Joining the Motoneuron team in June 2022, Morgan is investigating the implications of certain splicing defects in the establishment of the neuromuscular junction in DM1 and the development of novel cellular models for neuromuscular diseases.
Noémie Bérenger-Currias
Research Engineer (Inserm)
Noémie joined the Motoneurones team in May 2022. She works on the TREAD screening project, in collaboration with our partners Ksilink and LDC. In particular, she is characterizing the in vitro therapeutic potential of candidate pharmacological compounds for DM1.
Laurine Merriadec
Research Assistant (CECS)
Laurine joined the Motoneurons team in March 2023. She is working on the TREAD screening project, in collaboration with our partners Ksilink and LDC, in the characterization of the in vitro therapeutic potential of candidate pharmacological compounds for DM1.
Laetitia Aubry
Associate Professor (UEVE)
Leader of the Wolfram syndrome research project, Laetitia joined the Motoneurone team in 2022.
Sandra Pourtoy
Research Assistant (CECS)
Sandra arrived at I-Stem in 2015 and works on the Wolfram syndrome research project and was attached to the Motoneurons team in 2022.
Lise Morizur
Research associate (CECS)
Specialized in the development of 3D cellular models mimicking biological processes, Lise’s interest lies in the use of 3D muscles in vitro to study myopathies.
Hugo Lenoir
Doctoral student (UMR861)
Hugo completed his M1 and M2 internships in the Motoneuron team in 2022 and 2023. He started his PhD in October 2023, working on the therapeutic potential of extracellular vesicles in Duchenne muscular dystrophy.
Caroline Hermitte
Doctoral student (UMR861)
Caroline joined the motoneuron team for her Master 2 internship in January 2023, working on splicing defects involved in neuromuscular junction establishment in Dystrophic Myotonia Type 1 (DM1). Since November 2023, she has been a PhD student at I-Stem, working on the development of neuromuscular organoids in the pathologies of Dystrophic Myotonia Type 1 and infantile spinal muscular atrophy (SMA).
Minh Ngoc Khong
Phd student
Minh completed her M1 and M2 internships at I-Stem in 2023 and 2024. Since November 2024, she has been pursuing a PhD within the Motoneuron team, working on a 3D muscle cell model for DM1.
Myriam Mederic
Assistant Engineer (Inserm)
Myriam joined IStem in July 2025 following a mobility program. As an assistant engineer, she works with Sandrine Baghdoyan on designing various genomic tools for the team using CRISPR technology.
Mathurin Bodin
Phd student
After joining the Motoneurone team in February 2025 as part of his Master’s 2 internship, Mathurin has been pursuing his PhD there since October 2025. Using multi-omic approaches on organoid models, he is studying the development of the neuromuscular system in spinal muscular atrophy (SMA).
Naïma Sedrati
Engineer
Naïma joined the Motoneuron team in January 2025 as a Master’s 2 intern, and has been working there since October 2025 as a research engineer on the development of a DM1 functional screening test to identify DMPK gene regulators.
Moubani Das
Phd Student
Moubani joined in Feb 2026 as an MSCA doctoral fellow under ENTRY-DM network (EU funding). Her PhD evaluates neuromuscular and cortical organoids as DM1 models to validate therapies and elucidate disease pathophysiology.
Lise Josset
Engineer
Collaborations
ANR PRCE : Collaboration to identify new pharmacological leads for Steinert’s Myotonia.
ANR PRC : This collaboration aims to better understand the physiopathological mechanisms involved in the development of amyotrophic lateral sclerosis.
ANR PRC : This collaboration aims to better understand the pathophysiological mechanisms involved in the neurological disorders associated with Dystrophic Myotonia Type 1.
ANR PRC : This collaboration aims to better understand the consequences of alternative splicing defects associated with Dystrophic Myotonia type 1.
SMA Europe : This collaboration aims to identify new therapeutic avenues for infantile spinal muscular atrophy.
eRARE ReCognition : This collaboration aims to understand the molecular mechanisms that may be involved in the beneficial response observée dans l’essai Optimistic sur la Myotonie Dystrophique de type 1. Ce projet européen implique une dizaine d’équipes européennes et du Canada.
Publications
Alternative Splicing of SORBS1 Affects Neuromuscular Junction Integrity in Myotonic Dystrophy Type 1.
01 December 2025
Journal of cachexia, sarcopenia and muscle
Magnetic Bioprinting and Actuation of Stretchable Muscle Tissue.
01 January 2026
Advanced healthcare materials
Tale of mitochondria and mitochondria-associated ER membrane in patient-derived neuronal models of Wolfram syndrome.
01 September 2025
Neural regeneration research